Running Projects
Summary : Atto (1 as = 10−18 s) and few femtosecond (1 fs = 10−15 s) optical pulses allow exciting and probing electrons and nuclei in molecules on their intrinsic timescales. Their broad energy bandwidth of 0.5 to few eV leads to exciting selectively superpositions of several excited electronic states, building electronic coherences, which opens the way to shape the correlations between electronic and nuclear motions that are the heart of bond breaking and bond making at the excitation step. The main goal of MOLECOH is to develop a rational approach for designing control schemes of chemical reactivity that exploit photoexcited electronic coherences. Our aim is to design control schemes based on selectively exciting electronic coherences to control the correlation between electronic and nuclear motions on an ultrafast time scale before significant dephasing has occurred. Exciting specific coherences in molecules will be achieved by tuning the parameters of the attopulse: Its carrier frequency, duration, polarization and carrier envelope phase. Fully quantal dynamics for several coupled electronic states and few nuclear degrees of freedom will be implemented. On the fly semiclassical simulations in full dimensionality of the nuclear degrees of freedom will be used to identify those relevant nuclear coordinates that are essential for simulating the short time quantum dynamics. To enable a rational design of the control schemes, the entanglement of the vibronic wave packet will be quantified as a function of time, as well as the force it exerts on the nuclei.
• FNRS-FWO Excellence of Science project Tethered: ‘Keeping the best of weak bonds by tethering: a multi-scale, multi-technique approach to polymeric materials with controlled and predictable mechanical properties.’ 2022-2026 Coordinator: Charles-André Fustin (UCLouvain), Francoise Remacle : CoPI, leader WP4 ‘Modeling’, https://www.molsys.uliege.be/cms/c_8639112/en/eos-excellence-of-science-project-to-anne-sophie-duwez-and-francoise-remacle
Partners :
Françoise Remacle, Theoretical Physical Chemistry, University of Liege, Belgium
Hong-Guang Duan, Ningbo University, Ningbo, China
This project is a joint experimental and theoretical study of the wave packets dynamics at conical intersections. The experiments will be carried out in the group of Professor Hong-Guang Duan at the Ningbo University, China, and the theory-modeling part in the group of Professor Françoise Remacle at the University of Liège, Belgium. Experimentally, electronic, vibrational coherence and population dynamics in the vicinity of conical intersections will be investigated by Prof. Duan by 2 dimensional (2D) electronic spectroscopy using sequences of tailored ultrashort femtosecond laser pulses. The group of Prof. Remacle will be in charge of the modeling of the response to sequences of laser pulses and of the computation of the 2DES spectra. These simulations will be based on detailed electronic structure computation of the excited electronic states of the molecular systems, as well as their non adiabatic couplings. The 2D spectra will be computed from quantum dynamical simulations. Photosynthetic chlorophyll complexes will be investigated. Our project will shed light on the role of CI's and of vibronic coherences in energy and charge transfer processes in light harvesting complexes.
Previous Projects
International Projects
The main objectives of this proposal are to achieve two major research goals of ultrafast science:
1. Creating and probing photo-induced charge migration dynamics of pure electronic origin on the attosecond to few femtoseconds time scales and studying the role of non-equilibrium charge distributions in inducing selectivity of chemical reactions in polyatomic molecules.
2. Achieving mode-selective chemistry in polyatomic molecules using intense ultrashort mid-infrared pulses.
Scientific Discovery through Ultrafast Materials and Chemical Sciences, Department of Energy, United States

European projects
-
Core member of the COST action ATTOCHEM (CA18222) : Attosecond Chemistry, 2019-2024.
https://attochem.eu/
-
Partner in the ULiege team of the EC master program : AMIS
https://amis-master.eitrawmaterials.eu/
We use the dynamic response of the designed QD arrays to implement novel paradigms for parallel information processing. The discrete quantal level structure of nanosystems provides a memory at room temperature. Input will be provided simultaneously to all the levels by broadband laser pulses and the dynamical response will implement the logic in parallel. Disorder and environmental fluctuations are not detrimental because controlled level broadening is essential for the simultaneous multidirectional optical readout at the macroscopic level.
The long term vision of COPAC is the application of atomic and molecular state resolved controlled quantum dynamic processes towards information processing. Within this our targeted breakthrough is a novel prototype device for parallel logic engineered to industry standards and with suitable compilers.
http://www.copac-fet.eu
http://cordis.europa.eu/projects/rcn/110508_en.html
www.multivaluedlogics.eu/
1. Fabrication and measurement of devices which are inherently low-power in switching operation at room temperature.
2. Theory of specific device implementations for each of those technologies to explain and validate the principles behind their low-power capabilities.
3. Design of architecture to enable the circuit operation of these technologies for overall low-power circuit operation. ...
www.tolop.eu
www.moloc.ulg.ac.be
www.moldynlo.ulg.ac.be
Wallonia-Brussels Federation
Porte-parole : Françoise Remacle.

MONACOMP proposes a theoretical and methodological approach towards realizing ultrafast controlled parallel information processing on the nanoscale at room temperature. The logic is performed as the quantum dynamical response of ensembles of molecular complexes or of nanoparticles (NP) to a sequence of short few cycle femtosecond laser pulses. The parallelism is made possible by the broad bandwidth in energy of short fs pulses, which allows exciting a large band of quantum states coherently and in parallel. The information encoded by the pump pulses is processed by the quantum dynamical coherent time evolution until the read out by a probe pulse. In MONACOMP, we encode logic variables in the elements of the density matrix of the quantum systems, which allows an ultraDENSE encoding : N2-1 (for a normalized density) classical variables can potentially be encoded in an N level quantum system. The physical realization of the ultra dense encoding in observables has been experimentally demonstrated using ultrafast pump-probe non linear three or more wave mixing 2D electronic spectroscopy (2DES) which has been implemented in the condensed phase at room temperature. 2DES provides a reading of the amplitudes of the coherences by probing the stimulated emission of an ensemble of molecular complexes or nanoparticles in specific phase matching spatial directions, which provides directly a macroscopic output signal. The targeted logic operations are the spectral decomposition of multivariate multivalued logic functions. The quantum dynamics will be computed by integrating the time dependent Schrödinger equation using excitonic Hamiltonians in the non perturbative limit, including explicitly the interaction with the pulses. The response of stoichiometric systems (molecules and small Au and CdSe clusters) will be contrasted with that of larger CdSe NP, for which the dispersion in sizes induces a dispersion in the electronic properties and in the 2DES signal.
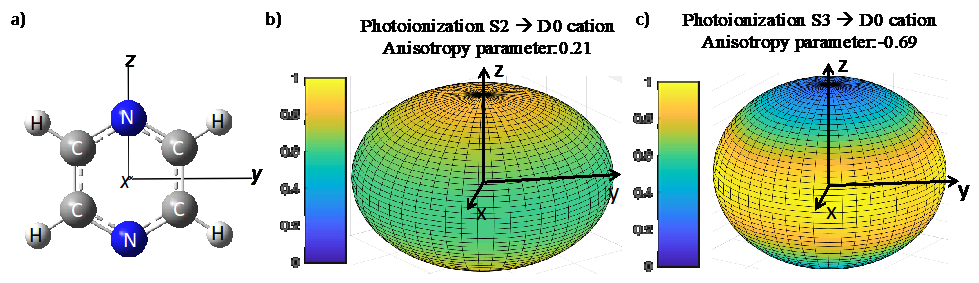
We aim at exploiting the complementary between the Tohuku and Uliège teams for developing the theoretical tools needed to better understand ultrafast photoinduced molecular dynamics in large systems. Since large molecules have a dense manifold of electronic states, it is challenging to describe accurately the nuclear and relaxation dynamics as well as to interpret pump-probe experiments monitoring these nuclear rearrangements. When the delayed probe pulse ionizes a wavepacket delocalized on several electronic states close in energy, the time-dependent photoelectron spectrum has a complex structure. Thus the contribution of each state needs to be disentangled to follow the wavepacket motion during the chemical reaction.
Our first goal is to implement a code to compute angularly resolved photoionization cross-sections and time-dependent photoelectron spectra, which depends on the electronic and nuclear dynamics. So it is important to accurately describe all the quantum effects that may arise, such as the interferences between wavepackets evolving on different electronic states; a situation often encountered in molecules with dense sets of strongly coupled states.
The second goal is to develop semi-classical approaches and make the approximations necessary to apply these methods to molecular systems while still capturing the essence of quantum effects. We will first expand the wavepacket in term of Gaussians and compute the electronic structure on the fly. We will benchmarked these simulations to quantum simulations on a grid for 2 and 3D systems. Then we will compare the ‘on the fly’ grid method for quantum dynamics that we recently proposed to the Gaussian based one proposed by our Tohoku partners. We will focus on the photoinduced dynamics of pyrazine before investigating larger systems.
Overall this joint project will provide crucial methodological developments needed for the interpretation of photochemical experiments of the large molecular systems that are nowadays investigated.
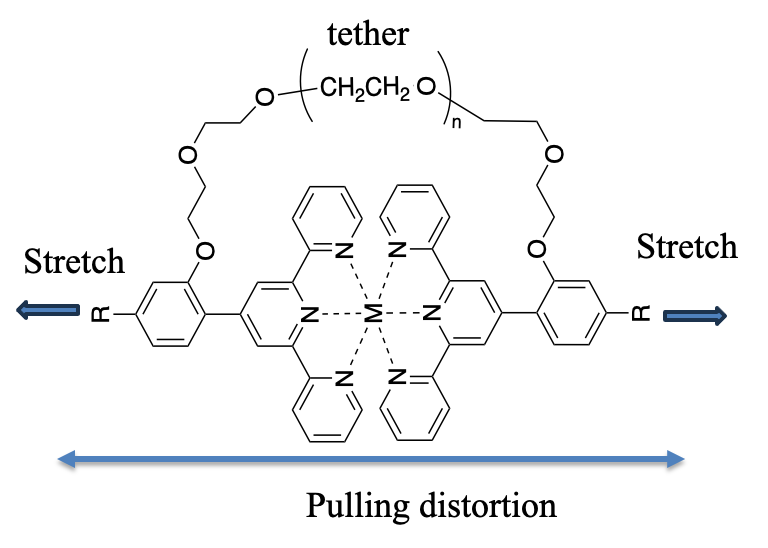
MECHANOCHEM will investigate the mechanical stability of mechanophores and the mechanical reversibility of click reactions at the molecular level, by a synergistic approach combining AFM-based SMFS and theory and modelling.
SMFS will be used to measure the mechanical force required to break open (i) a Diels–Alder-based mechanophore with different geometries and (ii) click linkages of three TAD-based adducts. Standard force spectroscopy experiments will be realized, as well as dynamic and force-clamp experiments. In standard force spectroscopy, a ramp of force is applied while the extension of the molecule is recorded. Dynamic force spectroscopy consists in measuring the rupture forces at different loading rates. Force clamp experiments enable to realize mechanical and kinetics measurements at a constant force. The force is clamped while the movements of the molecule are monitored as a function of time. Even if SMFS is very demanding and never a routine technique, standard and dynamic force spectroscopy are relatively easily implemented. Force clamp spectroscopy is much more challenging and delicate. All these experiments will allow us to determine the force required to break open the adducts and the time they can resist to a constant force.
The main objectives of the theory and modelling research effort are to provide an understanding of the regio and stereo selectivity that can be achieved in Diels–Alder reaction studied experimentally by single-molecule force spectroscopy using the strength and the direction of the applied external force. The analysis of the experimental results will be supported by theoretical modelling at the molecular level of the effect of the external mechanical force on the geometry of the mechanophores. Both static and dynamical methodologies will be implemented on the systems studied experimentally. In the static approach, we will implement the isometric (CoGEF) and isotensional approaches. As discussed above, these two approaches are complementary and lead to identical results for the stationary points of the force modified potential. For regions of non stationarity, they provide complementary information on the response of the system. The distribution of the strain energy in the different normal modes will be analysed using the harmonic approximation.[62] The features of the molecular orbitals along the minimum energy reaction path, i.e. their bonding and antibonding character, will be analysed. The information obtained using the stationary approaches will allow to characterize how the reaction mechanisms (energetics and kinetics) connecting the reactants and the different Diels–Alder products. We will then complement these studies by dynamical simulations that allow for including temperature effects.
In parallel, we also plan to investigate a purely theoretical aspect of the computation of the response to the mechanical force.
The experimental realization of ultrashort optical pulses opens new avenues towards photoinduced molecular reactivity. The reason is that a short, subfs to few fs optical pulse has a broad energy width, ranging from 0.5 eV to 1-2 eV, which enables a coherent excitation of several excited electronic states. Such an excited state is an electronic density that is not in equilibrium with the nuclei. This is a behavior not allowed in the Born Oppenheimer approximation. We propose to investigate the novel prospect of enabling and controlling the formation of new bonds. Ultrashort pulses were so far shown to lead to selectivity in bond breaking of diatomics and small polyatomic molecules. Our goal is to investigate theoretically ultrafast photoinduced bond making. We will study pericyclic isomerization and cyclo addition reactions. These reactions typically involve limited electron migration along the nuclear backbone accompanied by small displacements of the nuclei. Our rationale is that attopulses are particularly well suited for inducing these reactions because of the control they provide on the spatial and temporal localization of the excited coherent non equilibrium electronic density. We envision that by a suitable ultrashort excitation, it could be possible to localize in a controlled way a sufficient fraction of electron density where the bond needs to be made, thereby triggering a nuclei response that will trap the electron density between the targeted nuclei and lead to ultrafast bond formation. We will focus on the role of coherences, electronic and vibronic, to control the forces on the nuclei that induce bond making and assess if electronic coherence can favor a concerted ultrafast mechanism for the reaction. We will further investigate how the stereoselective Woodward-Hoffmann rules based on orbital symmetry and on the excitation of a single optically active electronic state are modified for a coherent superposition of several electronic states.
Our project aims to develop the theory and modeling tools necessary for controlling reactivity in molecules and nanostructures, based on the attosecond excitation of non-equilibrium states. The project focuses on non-equilibrium, multi-timescale, electron and ion dynamics, going all the way from the shortest time scale attainable (the atto to few fs (or subfs) range needed to excite a non-stationary state for the electrons) up to the time scale describing the targeted physicochemical outcomes of the excited system, which is longer by up to four orders of magnitude when the atoms have to move substantially. ...
Extreme miniaturization is made necessary by the steadily increasing requirements of information technologies and smart sensors, which are expected not only to detect and quantify the presence of a substance but also to handle in situ, in a simple way, the information obtained [1-5]. The implementation of physical systems able to handle the information at the nanometric and molecular scales calls for a strong basic effort aiming at a better understanding of the forces governing at those scales the interactions between the constitutive subunits and between the latter and the substrate. ...